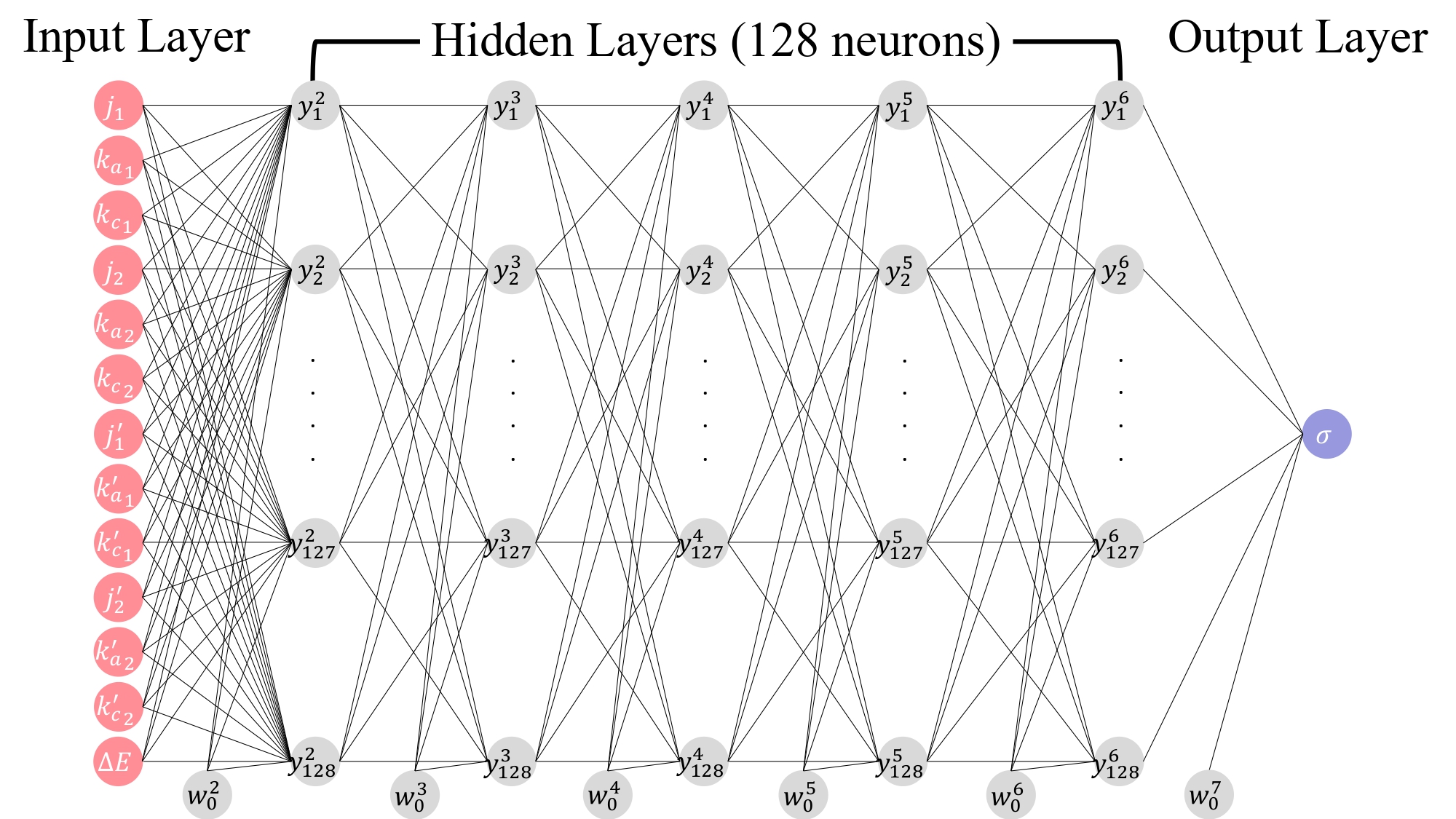
Quantum control of chemical reactions and inelastic scattering of molecules remain at the forefront of modern theoretical and computational chemistry. Quantum control can be achieved through quantum interference effects or quantum state preparations of molecular states, e.g., controlling alignment or orientations of the reactant molecules, generally referred to as stereodynamics. These effects are amplified at low temperatures where quantum effects dominate. Recent advances in molecular cooling and trapping technologies have also led to renewed interest in inelastic and reactive collisions of atom-molecule and molecule-molecule systems. Indeed, collisions of cold and ultracold diatomic molecules are an active area of experimental and theoretical research due to their applications to quantum sensing, ultracold chemistry, quantum computing, and quantum information processing. Although, diatomic species such as CaF, SrF, RbCs, KRb, NaK, etc. are preferred molecules for such applications, their small rotational constants lead to high densities of states making rigorous quantum calculations of diatom-diatom collisions involving these molecules computationally intractable. Therefore, lighter diatomic molecules, such as H₂ and its isotopologues, are generally preferred to benchmark theoretical studies against experiments. These systems are characterized by large rotational constants and low densities of states, thus requiring only a small number of orbital angular momentum partial waves to yield converged cross sections in the micro-kelvin (ultracold) range of collision energies.
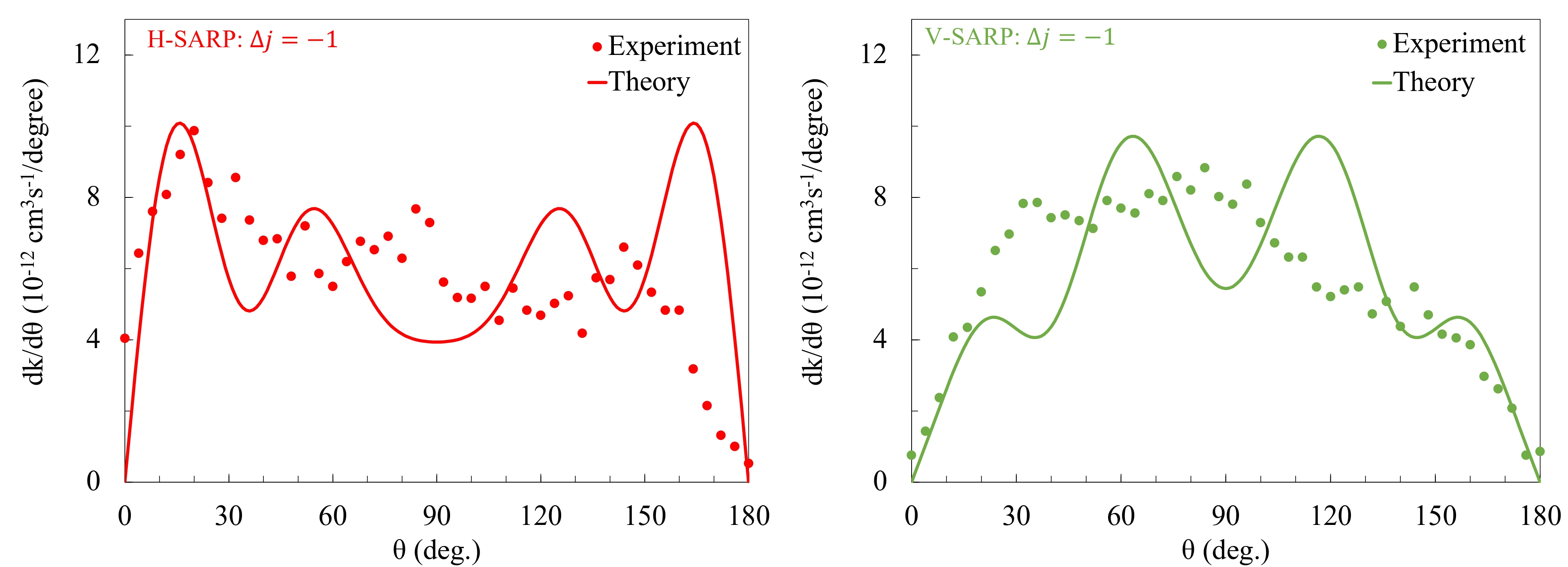
Prof. Richard N. Zare and his research group at the Stanford University have recently developed a coherent optical technique, called Stark-induced adiabatic Raman passage (SARP), to study quantum controlled cold collisions of light molecular systems, such as H₂, and its isotopologues. They conducted experiments on lighter molecular systems, starting from HD+H₂, HD+He and HD+D₂, D₂+He, D₂+Ne and D₂+D₂ using the new technique SARP. Their objective was to explore quantum control of molecular collisions in cold collision energy regime, particularly near 1 K. The quantum control is achieved by preparing the alignment of the molecular bond-axis relative to the initial velocity vector. The experiments can prepare parallel alignment, which is called H-SARP or perpendicular alignment, which is called V-SARP. The choice of light molecules such as H₂, HD, and D₂ restricts the number of partial waves to a few, typically below 𝓁=5, mostly, 𝓁 =0, 1 & 2. The experiments essentially created a new sub-field of quantum control of molecular collisions and chemical reactions through quantum state preparation & molecular alignment.
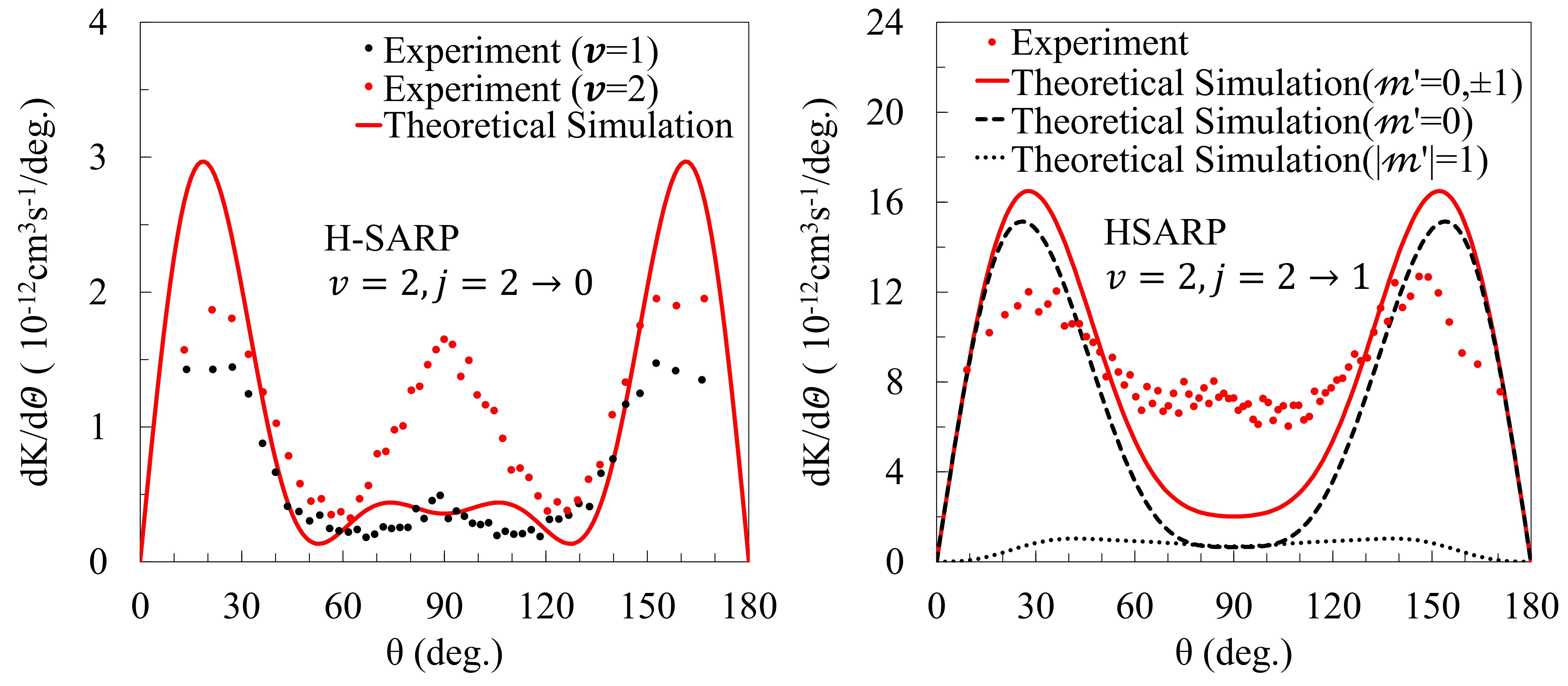
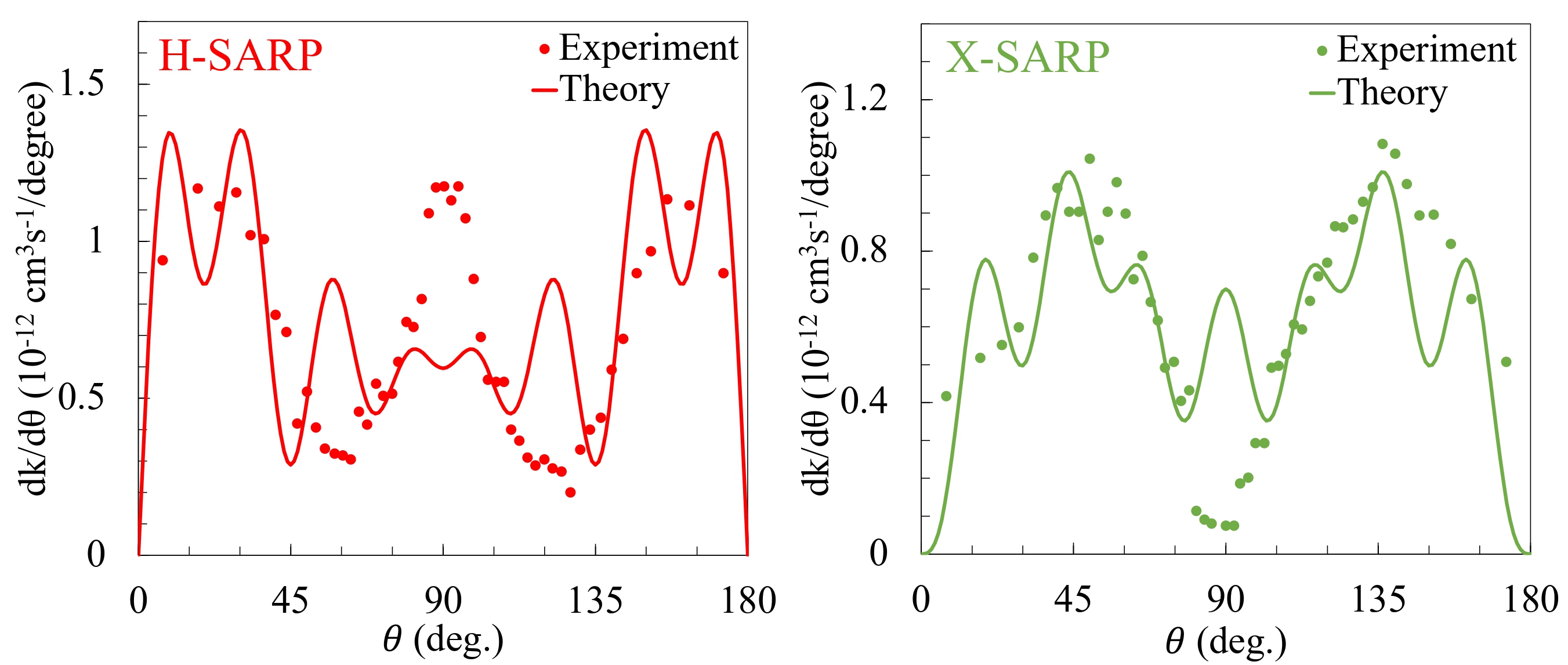
We conducted theoretical simulations of stereodynamic controlled cold collisions of HD+D₂, HD+He & D₂+He, and D₂(𝑣 = 4)+Ne to correctly interpreting experimental results and also to gain insights on molecular interactions and thus chemical reactions as well at a fundamental quantum level. The experiments involve an average over a broad range of collision energies, from nearly zero kelvin to a few kelvin; and thus the experimental results are not energy resolved. Analysis of experimental results relied on numerical fitting of the experimental data to a differential rate constructed from possible outgoing partial waves. This often led to incorrect interpretation of experimental results in terms of specific partial-waves that control the angular distribution. We conducted theoretical investigations to correctly identify specific partial-waves that control the angular distribution and explains the experimental outcome.